Cerebellar Paraganglioma: A Case Report and Literature Review
Dr. Abass Adam, Dr. Abdul-Aziz Mahama, Dr. Dickson Bandoh,
Dr. Munira Amadu, Dr. Hermas Saamaalme Paaga,
Dr. Okasha Issaka, Dr. Reuben Biitian Duti, Dr. Malcolm Mambuoraa Dery*
*Corresponding author: Dr. Malcolm Mambuoraa Dery, Neurosurgery Unit, Department of Surgery, Tamale Teaching Hospital, Tamale, Ghana; Email: malcolmdery135@gmail.com
DOI: 10.37722/JSOTA.2026103
Abstract
Background: Paragangliomas are rare neuroendocrine tumors that arise from extra-adrenal paraganglia. Primary cerebellar paragangliomas are exceptionally uncommon and may mimic other posterior fossa tumors, making diagnosis challenging.
Case Presentation: A 44-year-old male presented with a one-year history of progressive headaches and bilateral visual loss. CT and MRI identified a contrast-enhancing mass in the left cerebellar hemisphere crossing the vermis and later causing obstructive hydrocephalus. A ventriculoperitoneal shunt was inserted, followed by suboccipital craniotomy and C1 laminectomy for safe maximal resection of a solid-cystic, highly vascular lesion. Histopathology revealed a Zellballen pattern with S-100-positive sustentacular cells, confirming cerebellar paraganglioma. Postoperatively, headaches resolved, while blindness and ataxia persisted; the patient remained clinically stable at six-month follow-up.
Conclusion: Cerebellar paraganglioma is a rare differential diagnosis for posterior fossa masses. Diagnosis requires correlation of multimodal imaging, intraoperative findings, histopathology, and immunohistochemistry. Maximal safe surgical resection remains the cornerstone of management, particularly given the vascularity and diagnostic complexity of these lesions.
Keywords
Keywords: Cerebellar paraganglioma; Posterior fossa tumor; Zellballen; Neurosurgery; Case report.

Watch the Article in Motion
Intoduction
Paragangliomas are uncommon neuroendocrine neoplasms arising from paraganglionic tissue distributed throughout the body. Within the central nervous system, they most frequently arise in the jugulotympanic and cerebellopontine angle (CPA) regions, while primary cerebellar origin is exceedingly rare. Histologically, paragangliomas are characterized by nests of chief cells arranged in a Zellballen pattern and surrounded by S-100-positive sustentacular cells. These tumors may demonstrate significant vascularity and can radiologically mimic other posterior fossa neoplasms such as hemangioblastoma, metastasis, and ependymoma.
Maximal safe surgical excision remains the preferred treatment strategy, while radiotherapy is generally reserved for recurrent, residual, or unresectable disease. We report a rare case of cerebellar paraganglioma managed surgically at our institution and review relevant literature regarding diagnostic challenges, imaging characteristics, pathology, and management.
Literature Review
Intracranial paragangliomas are rare tumors, with the majority arising from the jugulotympanic region or cerebellopontine angle. Primary intraparenchymal cerebellar paragangliomas remain exceptionally uncommon, with only isolated case reports and small case series documented in literature. Prayson et al. described a pediatric cerebellar paraganglioma demonstrating the classic Zellballen architecture with S-100-positive sustentacular cells. Similarly, Chen et al. reported a pediatric cerebellar lesion with radiologic features initially suggestive of hemangioblastoma, highlighting the diagnostic overlap associated with these tumors.
Reports involving cerebellopontine angle and posterior fossa paragangliomas have emphasized symptoms related to raised intracranial pressure, cranial neuropathies, hydrocephalus, and cerebellar dysfunction. Akhtar et al. described a cerebellopontine angle paraganglioma presenting with hydrocephalus and cranial nerve deficits, findings comparable to the obstructive hydrocephalus observed in our patient.
Several reviews have further emphasized the importance of histopathological and immunohistochemical confirmation because imaging findings frequently overlap with hemangioblastoma, ependymoma, meningioma, and metastatic lesions. The characteristic Zellballen pattern, combined with positive staining for neuroendocrine markers and S-100-positive sustentacular cells, remains essential for definitive diagnosis.
Additionally, hereditary syndromes involving SDHx mutations, von Hippel-Lindau syndrome, MEN2/RET mutations, and neurofibromatosis type 1 have been associated with paragangliomas, particularly in younger patients and multifocal disease. These associations underscore the importance of long-term surveillance and consideration of genetic counseling in selected patients.
Case Presentation
A 44-year-old male presented to the Neurosurgery clinic of Tamale Teaching Hospital in June 2024 with a one-year history of progressive bilateral blurred vision and global headaches rated 7/10 on the numeric pain scale. He was alert and oriented with a Glasgow Coma Scale score of 15/15. Motor examination demonstrated normal power in all muscle groups with intact sensation. Cranial nerve examination was initially unremarkable apart from visual impairment.
Initial cranial CT demonstrated a hypodense lesion within the left cerebellar hemisphere extending across the vermis with surrounding edema and contrast enhancement. A cerebellar tumor, likely hemangioblastoma, was initially suspected, and MRI plus surgical intervention were advised. However, the patient defaulted follow-up because of financial constraints.
Three months later, he returned with worsening headaches and severe visual deterioration. Repeat CT imaging demonstrated obstructive hydrocephalus secondary to mass effect from the posterior fossa lesion. A ventriculoperitoneal shunt was inserted through the right Kocher’s point with subsequent resolution of headaches.
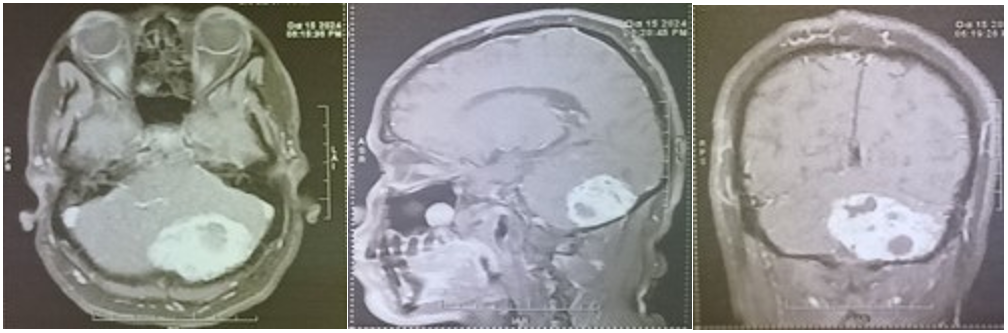
Four months later, reassessment demonstrated persistent complete bilateral blindness and cerebellar ataxia, though consciousness and motor examination remained intact. MRI demonstrated a predominantly hypointense lesion on T1-weighted imaging involving the left cerebellar hemisphere and vermis, with heterogeneous post-contrast enhancement and mixed solid-cystic components. T2-weighted and FLAIR sequences demonstrated hyperintensity within the lesion, while DWI and ADC imaging further delineated tumor characteristics. Preoperative laboratory blood workout revealed erythrocytosis with otherwise normal coagulation and renal function profiles.
Imaging and Intraoperative Findings
Radiologically, the lesion demonstrated features suggestive of a highly vascular posterior fossa tumor. The lesion crossed the vermis and exerted significant mass effect on adjacent cerebellar structures, eventually causing obstructive hydrocephalus. Contrast-enhanced MRI demonstrated heterogeneous enhancement with both solid and cystic regions, features commonly reported in hemangioblastoma and other vascular posterior fossa lesions.
The patient subsequently underwent suboccipital craniotomy with C1 laminectomy for maximal safe resection. Intraoperatively, the tumor appeared highly vascular, brown-tan in color, and solid-cystic in consistency. The lesion was identified within the left cerebellar hemisphere extending into the vermis. Careful circumferential dissection using approximately 2 mm margins of normal tissue enabled en bloc resection while preserving surrounding neurovascular structures. Hemostasis was meticulously secured.
Histopathology and Immunohistochemistry
Gross pathological examination demonstrated a brown-tan specimen measuring 4.5 × 3.5 × 2.0 cm. Microscopic examination revealed polygonal chief cells with uniform round nuclei and finely granular chromatin arranged in the classic Zellballen pattern between delicate vascular channels.
Immunohistochemical evaluation demonstrated S-100 positivity within sustentacular cells surrounding the Zellballen nests, supporting the diagnosis of paraganglioma. The histopathological and immunohistochemical findings were critical in distinguishing the lesion from hemangioblastoma, metastatic neuroendocrine tumors, and ependymoma. Unlike hemangioblastoma, paraganglioma demonstrates characteristic sustentacular cell staining and neuroendocrine architecture.
Differential Diagnosis
Radiologic differential diagnoses for adult posterior fossa masses include hemangioblastoma, metastasis, ependymoma, medulloblastoma, and meningioma. The highly vascular nature and enhancement characteristics of cerebellar paragangliomas may closely resemble hemangioblastoma. Although diffusion characteristics and lesion location may aid radiologic assessment, definitive diagnosis relies on histopathological and immunohistochemical confirmation.
Management and Follow-up
Maximal safe surgical resection remains the treatment of choice for localized cerebellar paragangliomas and provides excellent local disease control. Adjuvant radiotherapy may be considered in cases of residual tumor, recurrence, or anatomically unresectable disease.
Postoperatively, the patient experienced resolution of headaches, although blindness and cerebellar ataxia persisted without development of new neurological deficits. At six-month follow-up, he remained clinically stable and continued outpatient review.
Long-term follow-up with serial clinical assessment and MRI surveillance is recommended because of the risk of recurrence, particularly in incompletely resected lesions. Patients with atypical presentations, younger age, or multifocal disease may benefit from genetic evaluation and counseling due to recognized associations with hereditary syndromes including SDHx mutations, VHL syndrome, MEN2, and NF1.
Conclusion
Cerebellar paraganglioma is an exceptionally rare intracranial neoplasm that may mimic more common posterior fossa tumors both clinically and radiologically. Its diagnosis requires careful integration of multimodal imaging, intraoperative observations, histopathology, and immunohistochemical analysis. The highly vascular nature and challenging posterior fossa location make maximal safe surgical resection both diagnostically and therapeutically significant. This case contributes to the limited literature on cerebellar paragangliomas and highlights the importance of early diagnosis, multidisciplinary management, and long-term postoperative surveillance.
Declarations
Ethics approval and consent to participate: Ethical approval was waived at our institution, the Tamale Teaching Hospital Ethical Review Committee (TTH ERC). The ethical review waives ethical approval of case reports as patient identification is not revealed and would not be harmful to the patient.
Consent for publication: ritten informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors
Refernces
- Prayson RA, Goldblum JR, Schochet SS. Cerebellar paraganglioma in a child. Ann Diagn Pathol. 2004;8(3):168–173.
- Akhtar N, Altaf R, Hussain I, Qazi AA. Cerebellopontine angle paraganglioma presenting with cranial neuropathies and hydrocephalus: A rare case report. Surg Neurol Int. 2024; 15:45.
- Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs. 4th ed. IARC; 2017.
- Chen X, et al. Primary cerebellar paraganglioma: Pediatric case and literature review. J Neurooncol. 2014;120(1):181–186.
- Mendenhall WM, Amdur RJ, Hinerman RW, Mancuso AA, Villaret DB. Head and neck paragangliomas. Cancer. 2004;101(3):635–644.
- Boedeker CC, Ridder GJ, Schipper J. Paragangliomas of the head and neck: Diagnosis and treatment. Familial Cancer. 2005;4(1):55–59.
- Asa SL, Ezzat S, Mete O. The diagnosis and clinical significance of paragangliomas in unusual locations. J Clin Med. 2018;7(9):280.