Giant Cell Bone Tumour of Skull Base in an 8-Year-Old Male
– A Rare Case Report
Dr. Abass Adam, Dr. Abdul-Aziz Mahama, Dr. Dickson Bandoh,
Dr. Munira Amadu, Dr. Hermas Saamaalme Paaga,
Dr. Okasha Issaka, Dr. Malcolm Mambuoraa Dery*
*Corresponding author: Dr. Malcolm Mambuoraa Dery, Neurosurgery Unit, Department of Surgery, Tamale Teaching Hospital, Tamale, Ghana; Email: malcolmdery135@gmail.com
DOI: 10.37722/JSOTA.2026104
Abstract
Background: Giant cell tumour of bone (GCTB) is a benign but locally aggressive neoplasm that rarely involves the cranial bones (<1% of cases). Pediatric skull base GCTBs are exceptionally rare.
Case Presentation: We report the case of an 8-year-old boy who presented with a one-year history of progressive left eye proptosis and complete loss of vision. Neuroimaging revealed a mass lesion involving the left frontal region with extension into the sphenoid wing, cavernous sinus, sella, and left orbit. The patient underwent surgical excision, with postoperative CT showing gross total resection of the intracranial component, but residual tumour persisted at the skull base. Histopathology confirmed a giant cell tumour.
Conclusion: This case highlights an unusual presentation of skull base GCTB in a child and adds to the limited literature on intracranial and cranial GCTB. Early recognition and multidisciplinary management are essential because of the tumour’s aggressive nature and potential for recurrence.
Keywords
Giant cell tumour, Skull base, Pediatric neurosurgery, Proptosis, Bone tumour

Watch the Article in Motion
Introduction
Giant cell tumour of bone (GCTB) is one of the most common benign but aggressive primary bone tumours. It arises from the non-bone-forming supportive connective tissue of the marrow and is characterized histologically by stromal cells interspersed with multinucleated giant cells. GCTB constitutes approximately 5% of all primary bone tumours and 20% of benign bone tumours. While commonly found in long bones such as the distal femur, proximal tibia, distal radius, humerus, and sacrum, cranial involvement is rare, accounting for less than 1% of cases. When present, the sphenoid and temporal bones are the most frequently affected sites, although cases involving the occipital, frontal, and ethmoid bones have also been reported.
GCTB usually affects skeletally mature individuals aged 15–40 years, with a slight female predominance. Its occurrence in children, especially at the skull base, is exceedingly rare, making this case a significant contribution to existing literature.
Case report
An 8-year-old boy presented with a one-year history of progressive protrusion of the left eye associated with complete loss of vision. There was no history of trauma, seizures, or systemic illness. On examination, he had marked proptosis of the left eye, ophthalmoplegia, and absent light perception.
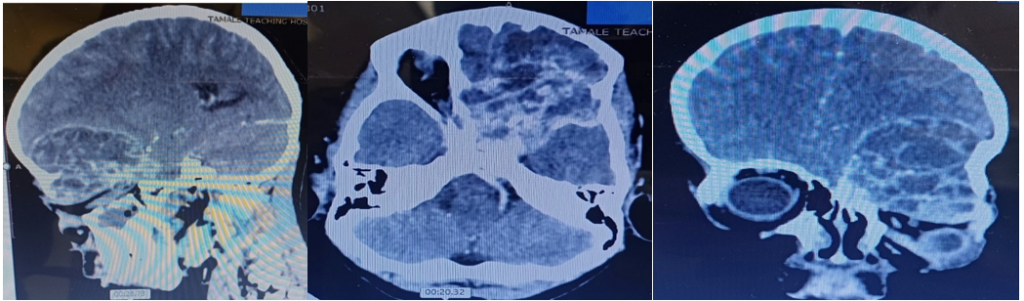
Imaging Findings: Preoperative CT and MRI revealed a large heterogeneous tumour involving the left frontal region with extension into the left sphenoid wing, cavernous sinus, sella, and orbit. Bone erosion of the sphenoid and skull base was evident.
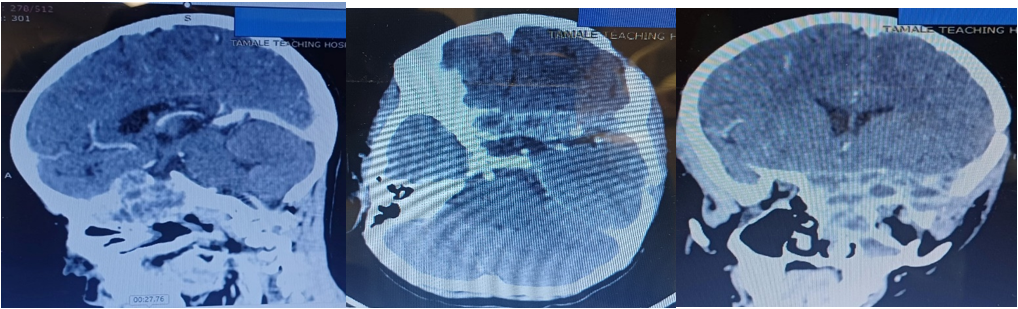
Surgical Intervention: The patient underwent craniotomy and microsurgical excision. Postoperative CT with contrast demonstrated complete removal of the intracranial tumour, although residual disease remained at the skull base.
Histopathology: Microscopic examination confirmed the diagnosis of giant cell tumour of bone, demonstrating numerous multinucleated osteoclast-like giant cells distributed within a background of spindle-shaped stromal cells.
Discussion
Clinical Presentation
Giant cell tumour of bone involving the skull base is exceedingly rare, particularly in the pediatric population. Only a limited number of pediatric skull base cases have been reported in literature. Similar cases reported by Sharma et al. and Patel et al. described symptoms related to mass effect and cranial nerve involvement, including proptosis, headache, and visual impairment, findings consistent with our patient’s presentation.
Patients with skull base GCTB commonly present with symptoms related to compression of adjacent neurovascular structures. In our patient, progressive unilateral proptosis, ophthalmoplegia, and complete visual loss were the dominant clinical features, likely resulting from orbital invasion and cavernous sinus involvement.
Imaging Findings
Radiologically, GCTBs typically appear as expansile osteolytic lesions with bone destruction and heterogeneous enhancement on CT and MRI. In this case, imaging demonstrated extensive involvement of the sphenoid wing, cavernous sinus, sella, orbit, and frontal region with associated skull base erosion. These findings are consistent with previously reported skull base GCTBs. However, imaging appearances are not pathognomonic and may overlap with aneurysmal bone cysts, fibrous dysplasia, chordoma, chondrosarcoma, eosinophilic granuloma, and giant cell reparative granuloma.
Histopathology and Differential Diagnosis
Histopathological examination remains the gold standard for diagnosis. The tumour demonstrated numerous multinucleated osteoclast-like giant cells dispersed among mononuclear spindle-shaped stromal cells, consistent with GCTB. Differential diagnoses included giant cell reparative granuloma, aneurysmal bone cyst, and brown tumour of hyperparathyroidism. Unlike giant cell reparative granuloma, GCTB demonstrates a more uniform distribution of giant cells and a more aggressive pattern of bone destruction.
Surgical Management
Maximal safe surgical excision remains the cornerstone of treatment for skull base GCTB. However, complete resection is often challenging because of the tumour’s proximity to critical neurovascular structures such as the cavernous sinus, optic apparatus, and internal carotid artery. In the present case, gross total excision of the intracranial component was achieved, although residual disease remained at the skull base.
Adjuvant therapies such as radiotherapy and denosumab therapy may be considered in cases of subtotal resection, recurrence, or unresectable disease. Denosumab, a RANKL inhibitor, has demonstrated promising results in reducing tumour burden and controlling disease progression.
Prognosis and Postoperative Monitoring
Although histologically benign, GCTBs are locally aggressive tumours with a recognized risk of recurrence, particularly following subtotal excision. Recurrence rates reported in literature range from 20% to 50%, depending on the extent of resection. Malignant transformation and distant metastasis are uncommon but have been documented.
Long-term postoperative surveillance with serial neuroimaging is therefore essential for early detection of recurrence or progression of residual disease. Careful follow-up is especially important in pediatric patients because of the aggressive nature of skull base lesions.
Multidisciplinary Management
This case highlights the importance of a multidisciplinary approach involving neurosurgeons, neuroradiologists, pathologists, ophthalmologists, and radiation oncologists in the diagnosis, treatment planning, postoperative care, and long-term follow-up of rare pediatric skull base tumours.
Conclusion
GCTB of the skull base in children is exceptionally rare but should be considered in pediatric skull lesions presenting with aggressive features such as proptosis and cranial neuropathies. Multimodal treatment strategies and long-term postoperative monitoring may be necessary to achieve disease control and reduce recurrence risk. This case contributes to the limited literature on pediatric skull base GCTB and highlights the challenges associated with management in complex anatomical regions.
Declarations
Ethics approval and consent to participate: Ethical approval was waived at our institution, the Tamale Teaching Hospital Ethical Review Committee (TTH ERC). The ethical review waives ethical approval of case reports as patient identification is not revealed and would not be harmful to the patient.
Consent for publication: ritten informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- Bertoni F, Bacchini P, Staals EL. Giant cell tumor (osteoclastoma) of bone. In: WHO Classification of Tumours of Soft Tissue and Bone. IARC Press; 2002.
- Sharma MC, Arora R, Deol PS, Mahapatra AK, Mehta VS, Sarkar C. Primary giant cell tumor of the skull: A series of 10 cases and review of the literature. Journal of Neuro-Oncology. 2001;53(1):39–44.
- Patel A, Agarwal V, Menon R, et al. Cranial giant cell tumour: Case report and review of literature. Pediatric Neurosurgery. 2010;46(4):297–301.
- Boriani S, Bandiera S, Casadei R, et al. Giant cell tumor of the spine: A review of 209 cases with long-term follow-up from a single institution. European Spine Journal. 2012;21(1):1–10.
- Thomas DM, Skubitz KM. Giant-cell tumour of bone. New England Journal of Medicine. 2009;360(23):2363–2375.
- Xu H, Nugent D, Monforte HL, et al. Clinical, pathological, and molecular characteristics of giant cell tumour of bone involving the skull. Histopathology. 2019;74(6):873–883.
- Errani C, Ruggieri P, Asenzio MA, et al. Giant cell tumor of the extremity: A review of 349 cases from a single institution. Cancer Treatment Reviews. 2010;36(1):1–7.
- Balke M, Campanacci L, Gebert C, et al. Bisphosphonate treatment of aggressive primary, recurrent and metastatic giant cell tumour of bone. BMC Cancer. 2010;10:462.
- Chawla S, Henshaw R, Seeger L, et al. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: Interim analysis of an open-label phase 2 study. The Lancet Oncology. 2013;14(9):901–908.
- Ohba T, Kawamura N, Ikuta K, et al. Giant cell tumour of the sphenoid bone: A case report and review of the literature. Oncology Letters. 2017;14(6):7111–7116.